PacBio Iso-Seq data analysis
An Introduction to PacBio Iso-Seq data analysis.
First Post
PacBio Sequel Systems (Sequel I and Sequel II) have two modes, Continuous Long Reas (CLR) and Circular Consensus Sequencing (CCS). 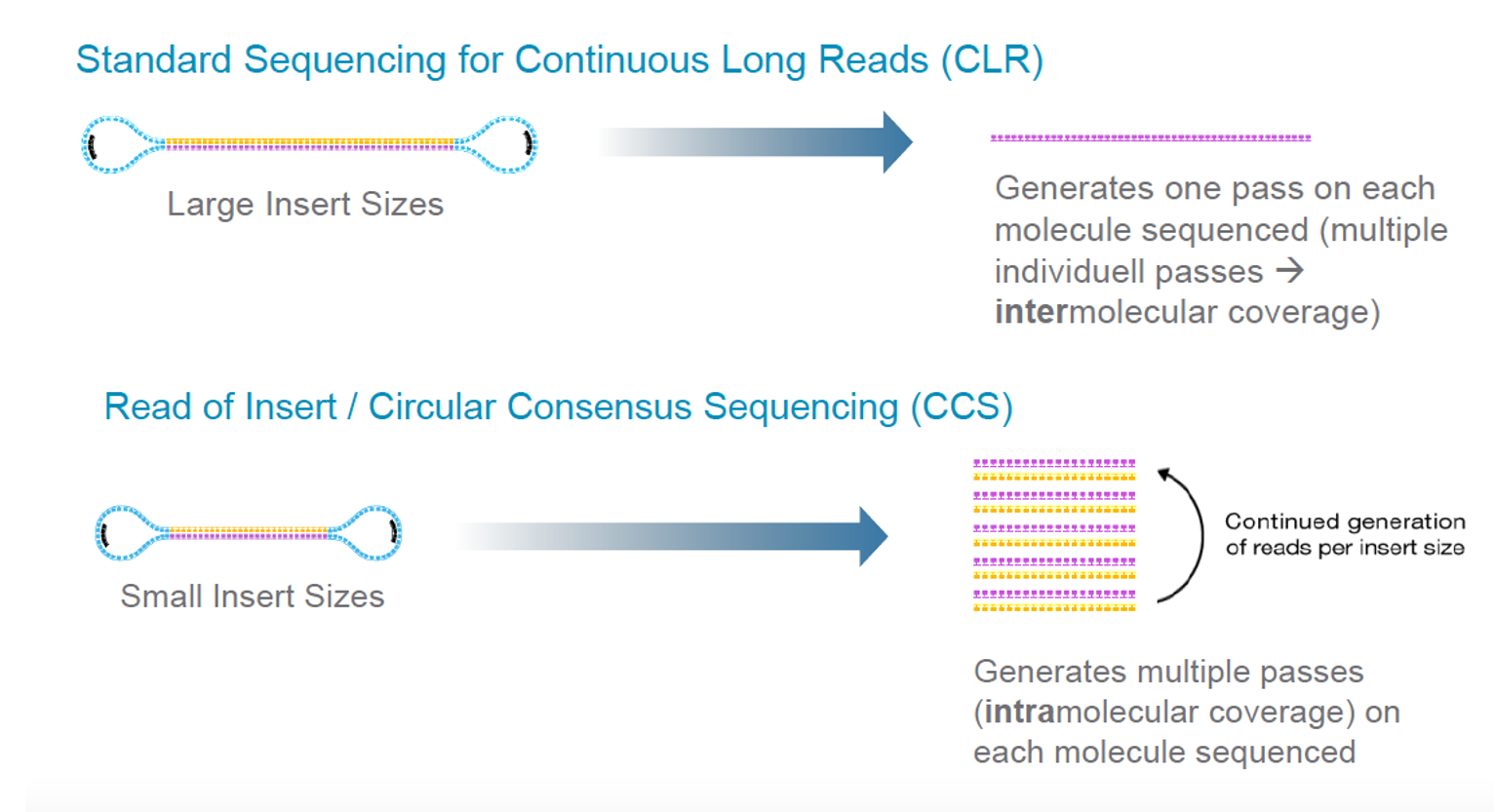
Software preparation
conda install -c bioconda pbccs # install CCS version 5.0 or above
conda install -c bioconda lima
conda install -c bioconda isoseq3 # install isoseq v3.4.0 or above
conda install -c bioconda pbcoretools # install dataset command
conda install -c bioconda bamtools
conda install -c bioconda minimap2
conda install -c bioconda samtools
Annotation preparation
wget -c https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_39/gencode.v39.annotation.gff3.gz
gunzip gencode.v39.annotation.gff3.gz
paftools.js gff2bed gencode.v39.annotation.gff3 > hg38.bigbed
Step1: Generate CCS (CCS reads)
If you don’t already have CCS reads, run ccs.
ccs [movie].subreads.bam [movie].ccs.bam
Options
--all # Emit all ZMWs
--min-rq # Minimum predicted accuracy in [0, 1]. using 0.99 by default.
--skip-polish # Only output the initial draft template (faster, less accurate)
Note that ccs run polish by default unless you don’t want it (use --skip-polish ), so additional polishing step (isoseq3 polish) is no longer needed.
A typical command to run ccs will be like this, as followed.
ccs [movie].subreadset.xml [movie].consensusreadset.xml --log-level INFO --report-json [movie].report.json --hifi-summary-json [movie].hifi_summary.json --log-file [movie].ccs.log --report-file [movie].report.txt --metrics-json [movie].zmw_metrics.json.gz -j 64
OR
ccs [movie].subreadset.bam [movie].ccs.bam --log-level INFO --report-json [movie].report.json --hifi-summary-json [movie].hifi_summary.json --log-file [movie].ccs.log --report-file [movie].report.txt --metrics-json [movie].zmw_metrics.json.gz -j 64
One important changes for ccs (>=v5.0.0) is that it has the --all mode. In this mode, ccs outputs one representative sequence per productive ZMW, irrespective of quality and passes.
Note that ccs is now running on the Sequel IIe instrument, transferring HiFi reads directly off the instrument. The on-instrument ccs version and also SMRT Link ≥v10 run in the –all mode by default.
But don’t worry, if you want to only use HiFi reads, ccs automatically generates additional files for your convenience that only contain HiFi reads:
- hifi_reads.fastq.gz
- hifi_reads.fasta.gz
- hifi_reads.bam
Step2: Barcode demultiplexer using LIMA (Full-length reads [FL reads])
We use the lima tool to remove the 5’ and 3’ cDNA primers
lima --isoseq --dump-clips [movie].ccs.bam primers.fasta [movie].fl.bam --peek-guess --log-file lima.log
Options
--isoseq # Activate IsoSeq mode
--dump-clips # Dump clipped regions in a separate output file <prefix>.lima.clips
--peek-guess # Try to infer the used barcodes subset.
lima identifies and removes the 5’ and 3’ cDNA primers. If the sample is barcoded, include the barcode as part of the primer.
Example 1: Following is the primer.fasta for the Clontech SMARTer and NEB cDNA library prep, which are the officially recommended protocols:
>NEB_5p
GCAATGAAGTCGCAGGGTTGGG
>Clontech_5p
AAGCAGTGGTATCAACGCAGAGTACATGGGG
>NEB_Clontech_3p
GTACTCTGCGTTGATACCACTGCTT
If there are more than two sequences in your primer.fasta file or better said more than one pair of 5’ and 3’ primers in Example 1, please use lima with –peek-guess to remove spurious false positive signal. If you provided only one pair of 5’ and 3’ primers in the primer.fasta, –peak-guess become unnecessary.
Output files will be called according to their primer pair. Example for single sample libraries:
[movie].fl.NEB_5p--NEB_Clontech_3p.bam
If multiple 5’/3’ pairs of primers are given, lima will output one <prefix>.<5p>--<3p>.bam for each pair. If you want to analyze all the demultiplexed FL reads together to increase transcript recovery (Example: Same species, different tissues), you must make a combined data set:
dataset create --type ConsensusReadSet combined_demux.consensusreadset.xml \
prefix.5p--barcode1_3p.bam \
prefix.5p--barcode2_3p.bam \
prefix.5p--barcode3_3p.bam ...
Step3: Remove PolyA Tail and Artificial Concatemers (full-length, non-concatemer reads[FLNC reads])
We use isoseq3 refine to remove polyA tail and artificial concatemers:
isoseq3 refine --require-polya [movie].5p--3p.bam primers.fasta [movie].flnc.bam
Options
--min-polya-length # Minimum poly(A) tail length. [20]
--require-polya # Require FL reads to have a poly(A) tail and remove it.
Analyze multiple the demultiplexed FL reads together, showed above.
isoseq3 refine --require-polya combined_demux.consensusreadset.xml primers.fasta flnc.bam
Output
Output The following output files containing full-length non-concatemer reads:
<movie>.flnc.bam
<movie>.flnc.consensusreadset.xml
An intermediate flnc.bam file is produced which contains the FLNC reads. To convert to FASTA format, run:
bamtools convert -format fasta -in flnc.bam > flnc.fasta
Now, flnc.fasta is the full-length, non-concatemer FASTA file you can use to align back to the genome for analysis!
Step4: Cluster FLNC reads and generate polished transcripts (HQ Transcript Isoforms)
isoseq3 cluster [movie].flnc.bam [movie].polished.bam --verbose --use-qvs
Options
--use-qvs # Use CCS QVs
Output
Note: Because the ccs was run with Polish, the isoseq3 cluster output is already polished! No additional polishing step is required. After completion, you will see the following files:
[movie].polished.hq.bam
[movie].polished.hq.bam.pbi
[movie].polished.lq.bam
[movie].polished.lq.bam.pbi
[movie].polished.hq.fasta.gz
[movie].polished.lq.fasta.gz
[movie].polished.cluster
[moive].polished.transcriptset.xml
Step5: Align to Genome
We currently recommend using minimap2 to align to the reference genome.
minimap2 -t 8 -Y -R "@RG\tID:Sample\tSM:hs\tLB:ga\tPL:PacBio" --MD -ax splice:hq -uf --secondary=no --junc-bed hg38.bigbed hg38.fasta polished.hq.fasta > aligned.sam
samtools sort -@ 8 -O BAM align.sam -o aligned.sort.bam
samtools index aligned.sort.bam
References
- Installing-and-Running-Iso-Seq-3-using-Conda
- Iso-Seq-Single-Cell-Analysis:Recommended-Analysis-Guidelines
- Best practice for aligning Iso Seq to reference genome: minimap2, deSALT, GMAP, STAR, BLAT
- LIMA barcoding
- Isoseq3 clustering(Generate transcripts by clustering HiFi reads)
- Isoseq3 deduplication (Generate transcripts by PCR deduplication (single-cell and UMIs))
- minimap2 manual
Enjoy Reading This Article?
Here are some more articles you might like to read next:
- From Bench to Bedside: A Practical Guide to Using Large Language Models in Medical Research
- Rethinking Bioinformatics Expertise in the Era of AI: A Newbie's Journey as a Custodian
- Mapping the Global Landscape of Cancer Research: Key Trends from the Nature Index
- Shattered Chromosomes: Understanding Chromothripsis and Genome Rearrangement Models
- Diffusion LLMs
- Nanopore raw data visualization using squigualiser
- Nanopore direct RNA data analysis using Dorado
- Modern commnad line tools powered by Rust
- Nanopore direct cDNA data analysis
- Hi-C data analysis