RNA-seq data analysis (strand issues)
Posted on September 21, 2016
I had been working on strand-specific paired-end reads from HiSeq lately and I had trouble mapping reads back to assembled transcripts using STAR as well as using RSEM to estimate transcript abundance.
Strand-specific protocol
| Library Type | Examples | Description |
|---|---|---|
| fr-unstranded | Standard Illumina | Reads from the left-most end of the fragment (in transcript coordinates) map to the transcript strand, and the right-most end maps to the opposite strand. |
| fr-firststrand (RF/R) | dUTP, NSR, NNSR | Same as above except we enforce the rule that the right-most end of the fragment (in transcript coordinates) is the first sequenced (or only sequenced for single-end reads). Equivalently, it is assumed that only the strand generated during first strand synthesis is sequenced. |
| fr-secondstrand (FR/F) | Ligation, Standard SOLiD | Same as above except we enforce the rule that the left-most end of the fragment (in transcript coordinates) is the first sequenced (or only sequenced for single-end reads). Equivalently, it is assumed that only the strand generated during second strand synthesis is sequenced. |
Bowtie and Tophat flags for strand-specific reads
Tophat uses –fr-firststrand for a library created by the dUTP method. This is stated clearly in the manual, so it is easy to understand. In contrast, Bowtie/Bowtie2 uses –fr, –rf, –ff to specify the orientation of paired-end reads.
--fr means the upstream read (/1) is from a forward strand and the downstream read (/2) is from a reverse strand.
--rf means the upstream read (/1) is from a reverse strand and the downstream read (/2) is from a forward strand.
--ff means both reads are from a forward strand.
But regarding to which strand the RNA fragment is synthesized from, this involves different strand-specific protocols. Thanks to the illustration figure (see below) from Zhao Zhang, we could see that for example dUTP method is to only sequence the strand from the first strand synthesis (the original RNA strand is degradated due to the dUTP incorporated), so the /2 read is from the original RNA strand.
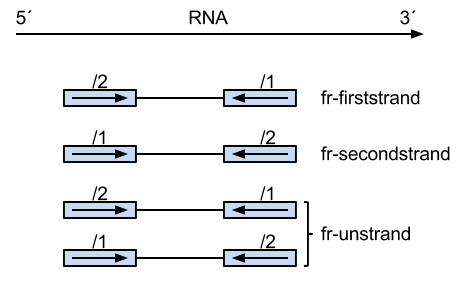
Summary of library type protocols (for Tophat/Bowtie)
With –fr flag, the upstream reads could be either /1 and /2, which is valid for unstranded reads. However, as you can see from the figure, the /1 reads from dUTP are from the reverse strand only. So, we need to tell Bowtie not to map the /1 reads to the forward strand. This behavior can be achieved by specifying –nofw flag.
Therefore, to run Bowtie on dUTP reads, the command should look something like this:
bowtie -S -X 1000 --fr --nofw bowtie-index -1 reads_R1.fastq -2 reads_R2.fastq > output.sam
To compare the results, I ran Bowtie on the same dataset (1M reads) with different flags specified.
| Flag | Mapped Reads | Unmapped Reads |
|---|---|---|
| –fr (default) | 82.57% | 17.43% |
| –rf | 0.17% | 99.83% |
| –ff | 0.00% | 100.00% |
| –fr –nofw | 82.18% | 17.82% |
| –fr –norc | 1.99% | 98.01% |
The number of reads mapped with –fr and –nofw is a bit lower than that from –fr alone. This is due to the fact that without –nofw flag, Bowtie maps /1 reads to both strands.
Note, with –norc flag, Bowtie will not attempt to map the /1 reads to the reverse strand.
But regarding to which strand the RNA fragment is synthesized from, this involves different strand-specific protocols. Thanks to the illustration figure (see below) from Zhao Zhang, we could see that for example dUTP method is to only sequence the strand from the first strand synthesis (the original RNA strand is degradated due to the dUTP incorporated), so the /2 read is from the original RNA strand.
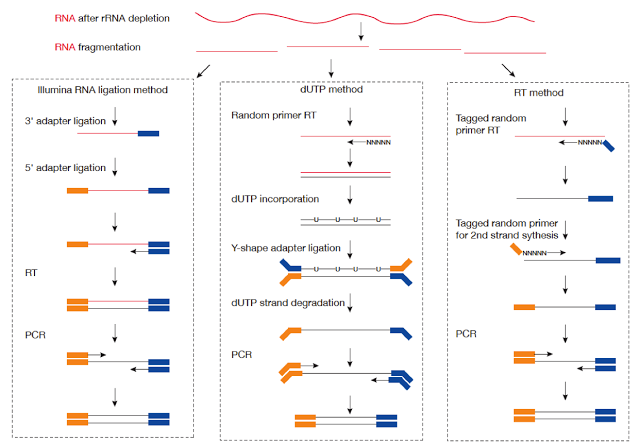
Strand-specific library protocols (Credit: Zhao Zhang)
Running RSEM on dUTP reads
RSEM has –strand-specific flag that will turn on Bowtie –norc flag, which will force Bowtie to try to map the /1 reads to the forward strand only. This is suitable for reads generated by the ligation method. Also, the –strand-specific flag is equivalent to setting –forward-prob flag to 1.
However, for dUTP reads, the –forward-prob flag should be set to 0 instead. It will specify –nofw flag, which forces Bowtie to map the /1 reads to the reverse strand. Consequently, the –strand-specific flag should not be used when –forward-prob is set to 0. The –forward-prob flag not only specifies –nofw and –norc for Bowtie, but also modifies the orientation parameters of RSEM model.
For example, the command should look something like this:
rsem-calculate-expression -p 8 --forward-prob 0 --paired-end sample_r1.fastq sample_r2.fastq index sample_output
Running HTseq-count on dUTP reads
dUTP-based libraries convey strand with read #2, so htseq-count –stranded=reverse will produce sense counts for such datasets. Using –stranded=yes would then yield anti-sense counts. Htseq-count was unfortunately designed at a time when non-dUTP methods were still popular, so its default stranded option is opposite of what’s now common.
For example, the command should look something like this:
htseq-count --format=bam --order=pos --stranded=reverse --type=gene --idattr=gene_id --mode=union Aligned.toTranscriptome.out.bam Homo_sapiens.GRCh38.82.gtf
Trinity Assembly
To run Trinity with dUTP reads, you need to use RF for –SS_lib_type flag, which means the /1 reads are from the reverse strand and the /2 reads are from the forward strand. Note that, RF flag in Trinity is not the same as –rf in Bowtie
Infer strand infomation
Actually we can use infer_experiment.py from RSeQC to infer how RNA-seq sequencing were configured, particulary how reads were stranded for strand-specific RNA-seq data, through comparing the “strandness of reads” with the “standness of transcripts”.
You don’t need to know the RNA sequencing protocol before mapping your reads to the reference genome. Mapping your RNA-seq reads as if they were non-strand specific, this script can “guess” how RNA-seq reads were stranded.
For pair-end RNA-seq, there are two different ways to strand reads:
- 1++,1–,2+-,2-+ (Ligation method)
- read1 mapped to ‘+’ strand indicates parental gene on ‘+’ strand
- read1 mapped to ‘-‘ strand indicates parental gene on ‘-‘ strand
- read2 mapped to ‘+’ strand indicates parental gene on ‘-‘ strand
- read2 mapped to ‘-‘ strand indicates parental gene on ‘+’ strand
- 1+-,1-+,2++,2– (dUTP method)
- read1 mapped to ‘+’ strand indicates parental gene on ‘-‘ strand
- read1 mapped to ‘-‘ strand indicates parental gene on ‘+’ strand
- read2 mapped to ‘+’ strand indicates parental gene on ‘+’ strand
- read2 mapped to ‘-‘ strand indicates parental gene on ‘-‘ strand
For single-end RNA-seq, there are also two different ways to strand reads:
- ++,– (Ligation method)
- read mapped to ‘+’ strand indicates parental gene on ‘+’ strand
- read mapped to ‘-‘ strand indicates parental gene on ‘-‘ strand
- +-,-+ (dUTP method)
- read mapped to ‘+’ strand indicates parental gene on ‘-‘ strand
- read mapped to ‘-‘ strand indicates parental gene on ‘+’ strand
For example : Pair-end strand specific:
infer_experiment.py -r hg19.refseq.bed12 -i Pairend_StrandSpecific_Human.bam
#Output::
This is PairEnd Data
Fraction of reads failed to determine: 0.0072
Fraction of reads explained by "1++,1--,2+-,2-+": 0.0487
Fraction of reads explained by "1+-,1-+,2++,2--": 0.9441So it is a strand-specific pair-end RNA-seq data using dUTP protocol.
In a nutshell
Alternative summary from Griffith Lab.
Thanks to Likit Preeyanon and Xianjun Dong for generously sharing ideas.