Nanopore direct RNA data analysis using Dorado
An Introduction to Nanopore direct RNA data analysis using Dorado. Dorado is a production basecaller from 2022, successor to Guppy.
Software preparation
# Install Dorado current version
wget -c https://cdn.oxfordnanoportal.com/software/analysis/dorado-0.7.1-linux-x64.tar.gz
tar zxvf dorado-0.7.1-linux-x64.tar.gz
# add dorado/bin to $PATH in .bashrc file
PATH=/path/to/dorado/bin:$PATH
# download dorado models
dorado download --model all
# install minimap2 and samtools
conda install -c bioconda minimap2 # paftools.js will be install automatically.
conda install -c bioconda samtools
Annotation preparation
wget -c https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_39/gencode.v39.annotation.gff3.gz
gunzip gencode.v39.annotation.gff3.gz
paftools.js gff2bed gencode.v39.annotation.gff3 > hg38.bigbed
Step1: Basecalling
GPU-based basecalling
# Adapters for RNA002 and RNA004 kits are automatically trimmed during basecalling
# Simplex basecalling
dorado basecaller <model> ./pod5/ > data.bam
# If you would like to keep the adapter sequences
dorado basecaller --no-trim <model> ./pod5/ > data.bam
# Duplex basecalling
dorado duplex sup ./pod5/ > duplex.bam
For duplex basecalling, it is suggested to use super-accurate basecaller(SUP) models, see ONT documentation
Options
--no-trim # do not trim the adapter or primer sequences
Step2: Convert BAM to FASTQ files
# For simplex basecalling
samtools view data.bam -d dx:0 | samtools fastq > data.fastq
# For dupllex basecalling
samtools view data.bam -d dx:0 | samtools fastq > data.fastq # simplex reads which don't have duplex offsprings
samtools view data.bam -d dx:1 | samtools fastq > data.fastq # duplex reads
samtools view data.bam -d dx:-1 | samtools fastq > data.fastq # simplex reads which have duplex offsprings
What is Simplex basecalling and Duplex basecalling?
Sometimes during ONT sequencing, after one strand of DNA finishes its trip through a pore, the other strand immediately follows, and there are two different ways that the basecaller can handle this. It can make a separate read from each strand’s signal, what ONT calls simplex sequencing. Or it can basecall both signals together to make a single read with higher accuracy, what ONT calls duplex sequencing. According to user’s experience1, about 15–20% of ONT read signals are part of a duplex pair.
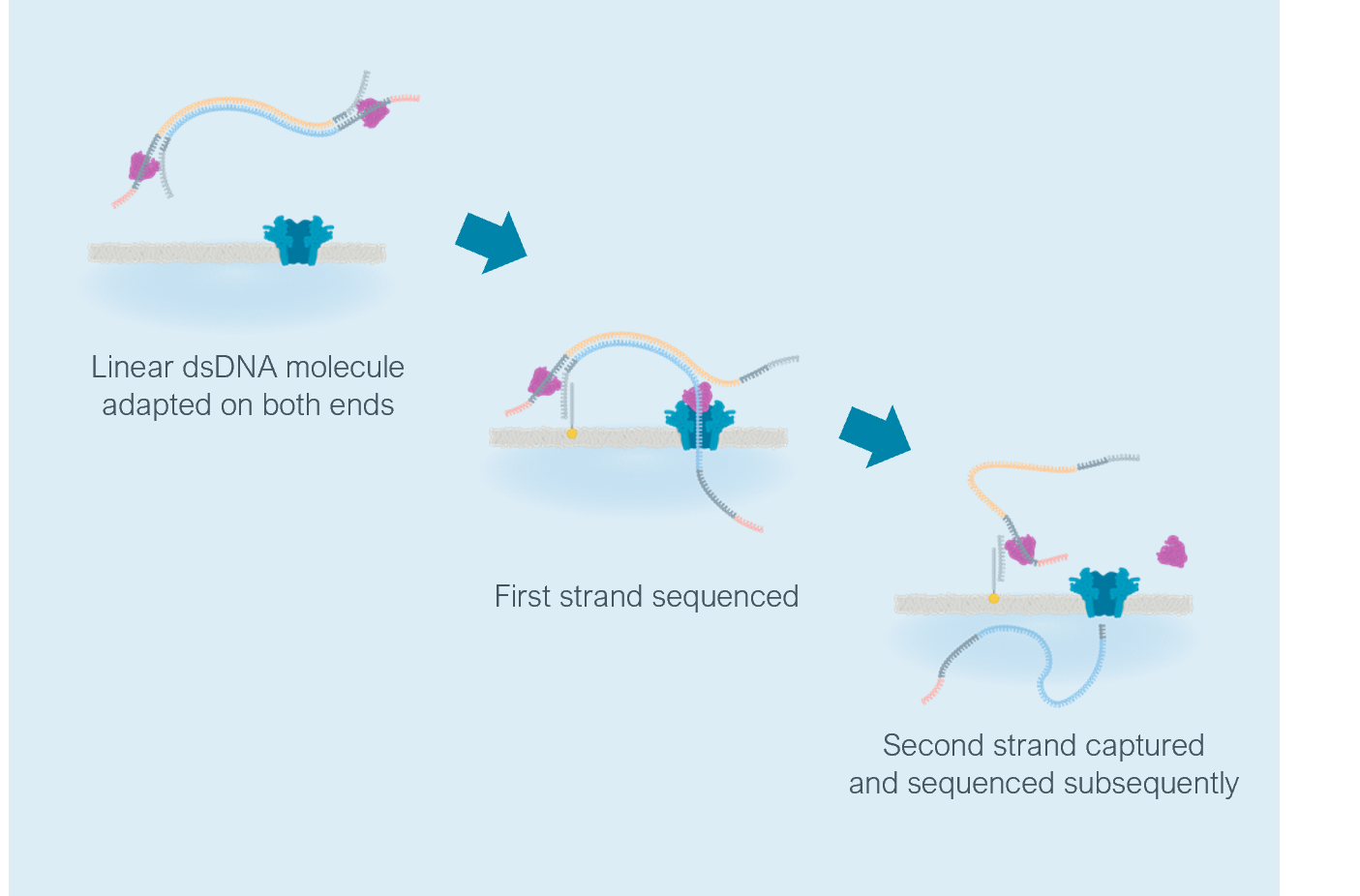
When using the duplex command, two types of DNA sequence results will be produced: ‘simplex’ and ‘duplex’. Any specific position in the DNA which is in a duplex read is also seen in two simplex strands (the template and complement). So, each DNA position which is duplex sequenced will be covered by a minimum of three separate readings in the output.
The dx tag in the BAM record for each read can be used to distinguish between simplex and duplex reads:
- dx:i:1 for duplex reads.
- dx:i:0 for simplex reads which don’t have duplex offsprings.
- dx:i:-1 for simplex reads which have duplex offsprings.
So there are two approaches one might use to basecall ONT sequencing runs:
- Simplex basecalling: Each signal is basecalled separately, whether or not it is part of a duplex pair. E.g. 1M read signals will generate 1M basecalled reads, and the read accuracy distribution will be monomodal.
- Duplex basecalling: Signals that are part of a duplex pair are basecalled together into duplex reads, and signals not part of a duplex pair are basecalled as simplex reads. E.g. 1M read signals with a duplex rate of 20% would generate 100k duplex reads and 800k simplex reads3, and the read accuracy distribution will be bimodal.
Step3: Aign to Genome
We currently recommend using minimap2 to align to the reference genome.
minimap2 -Y -t 8 -R "@RG\tID:Sample\tSM:hs\tLB:ga\tPL:ONT" --MD -ax splice -uf -k14 --junc-bed hg38.bigbed hg38.fasta data.fastq | samtools sort -@ 8 -O BAM -o aligned.bam -
samtools index aligned.bam
Optional: other analysis
You may try other tools for ONT data visualization and analysis2.
References
Enjoy Reading This Article?
Here are some more articles you might like to read next:
- From Bench to Bedside: A Practical Guide to Using Large Language Models in Medical Research
- Rethinking Bioinformatics Expertise in the Era of AI: A Newbie's Journey as a Custodian
- Mapping the Global Landscape of Cancer Research: Key Trends from the Nature Index
- Shattered Chromosomes: Understanding Chromothripsis and Genome Rearrangement Models
- Diffusion LLMs
- Nanopore raw data visualization using squigualiser
- Modern commnad line tools powered by Rust
- Nanopore direct cDNA data analysis
- Hi-C data analysis
- Nanopore direct RNA data analysis