Nanopore direct cDNA data analysis
An Introduction to Nanopore direct cDNA data analysis.
Software preparation
# Install Guppy CPU version
wget -c https://cdn.oxfordnanoportal.com/software/analysis/ont-guppy-cpu_6.4.6_linux64.tar.gz
tar zxvf ont-guppy-cpu_6.4.6_linux64.tar.gz
# Install Guppy GPU version
wget -c https://cdn.oxfordnanoportal.com/software/analysis/ont-guppy_6.4.6_linux64.tar.gz
tar zxvf ont-guppy_6.4.6_linux64.tar.gz
# add ont-guppy-cpu/bin to $PATH in .bashrc file
PATH=/path/to/ont-guppy-cpu/bin:$PATH
# install minimap2 and samtools
conda install -c bioconda minimap2 # paftools.js will be install automatically.
conda install -c bioconda samtools
# install pychopper
conda install -c epi2melabs -c conda-forge -c bioconda "epi2melabs::pychopper" # nhmmscan will be install automatically.
Annotation preparation
wget -c https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_39/gencode.v39.annotation.gff3.gz
gunzip gencode.v39.annotation.gff3.gz
paftools.js gff2bed gencode.v39.annotation.gff3 > hg38.bigbed
Step1: Basecalling
CPU-based basecalling
guppy_basecaller --input_path ./fast5 --save_path ./guppy_output --flowcell FLO-MIN106 --kit SQK-DCS109 --num_callers 16 --cpu_threads_per_caller 8 --compress_fastq --trim_strategy none
GPU-based basecalling
guppy_basecaller --input_path ./fast5 --save_path ./guppy_output --flowcell FLO-MIN106 --kit SQK-DCS109 --num_callers 16 --gpu_runners_per_device 80 -x "cuda:all" --compress_fastq --trim_strategy none
![]()
Do NOT Turn on trimming (setting --trim_strategy none ) during basecalling as it will remove the primers needed for classifying the reads!
Step2: Identify full-length Nanopore cDNA reads
In this step, Pychopper is used to identify, orient and trim full-length Nanopore cDNA reads. Pychopper can also rescue fused reads (chimeric reads).
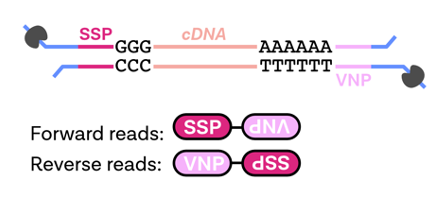
| Term | Description |
|---|---|
| SSP | strand-switching primer |
| VNP | anchored oligo(dT) VN primer |
Combine called FASTQ files
cat ./guppy_output/pass/*.gz > raw.fastq.gz
First round full-length cDNA reads identification with standard parameters using the default pHMM backend and autotuned cutoff parameters estimated from subsampled data:
pychopper -r report.pdf -k PCS109 -u unclassified.fq -w rescued.fq raw.fastq.gz full_length_output.fq
Second round full-length cDNA reads identification applied to the unclassified direct cDNA reads with DCS-specific read rescue enabled (parameter -x).
DCS109 can suffer from a specific reverse transcription artefact, which will lead to 2D-like reads with two VNP primers at the ends.
The -x DCS109 mode should be used on the unclassified reads only (while keeping the classified ones of course). This will classify the -VNP,VNP configurations as full length (also rescuing fused reads with this configuration, though they are not prevalent).
pychopper -r report_2.pdf -k PCS109 -x PCS109 -u unclassified_2.fq -w rescued_2.fq unclassified.fq full_length_output_2.fq
Full-length and rescued reads are merged and used for subsequent steps.
cat full_length_output.fq full_length_output_2.fq rescued.fq rescued_2.fq > full_length_cdna.fastq
Step3: Aign to Genome
We currently recommend using minimap2 to align to the reference genome.
minimap2 -Y -t 8 -R "@RG\tID:Sample\tSM:hs\tLB:ga\tPL:ONT" --MD -ax splice -uf -k14 --junc-bed hg38.bigbed hg38.fasta full_length_cdna.fastq > aligned.sam
samtools sort -@ 8 -O BAM align.sam -o aligned.sort.bam
samtools index aligned.sort.bam
References
Enjoy Reading This Article?
Here are some more articles you might like to read next:
- Rethinking Bioinformatics Expertise in the Era of AI: A Newbie's Journey as a Custodian
- Mapping the Global Landscape of Cancer Research: Key Trends from the Nature Index
- Shattered Chromosomes: Understanding Chromothripsis and Genome Rearrangement Models
- Diffusion LLMs
- Nanopore raw data visualization using squigualiser
- Nanopore direct RNA data analysis using Dorado
- Modern commnad line tools powered by Rust
- Hi-C data analysis
- Nanopore direct RNA data analysis
- Operation on BigWig Files